What is postmarket safety surveillance?
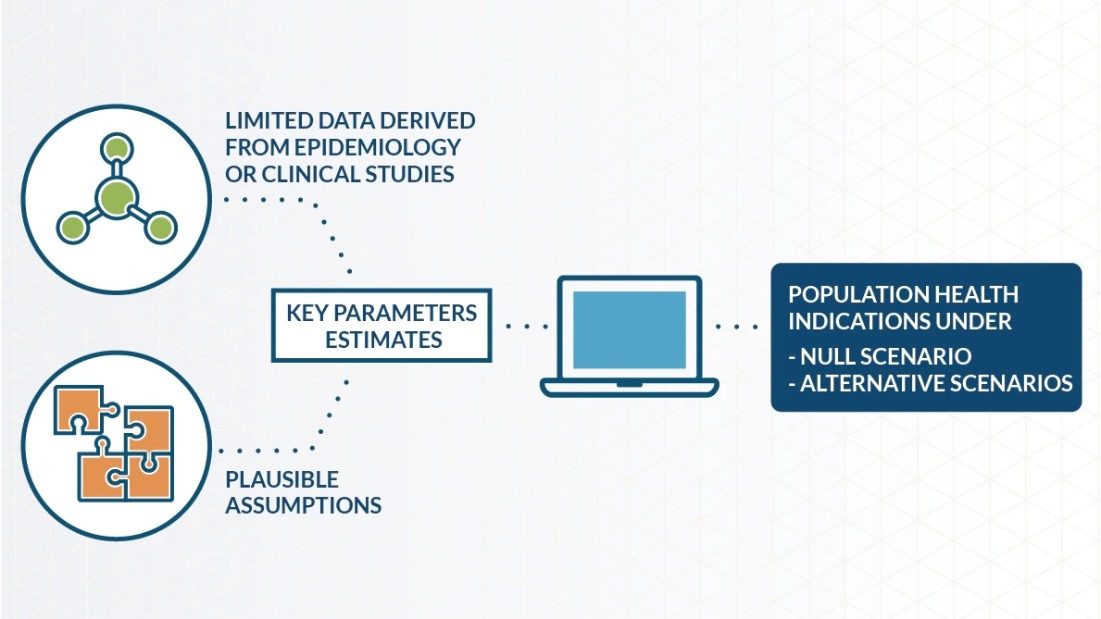
While premarket authorizations assess safety before product release, postmarket safety surveillance is vital for ongoing monitoring of safety concerns and is a key step in the assessment of smoke-free products. Regulatory agencies also play a vital role in evaluating safety data and ensuring necessary actions to mitigate risks. The U.S. Food and Drug Administration (FDA) defines postmarket surveillance as “… the active, systematic, scientifically valid collection, analysis, and interpretation of data or other information about a marketed device.”
At PMI, the safety and regulatory compliance of our smoke-free products is paramount. The goals of our postmarket safety surveillance include understanding the benefits and risk profile of our products, implementing measures to minimize risks to consumers, complying with local regulation, and ensuring that safety is taken into consideration throughout the entire product lifecycle.
PMI conducts its safety surveillance in alignment with pharmaceutical standards inspired by EU Good Pharmacovigilance Practices (GVP), strengthening the robustness of our processes for clinical and postmarket research. Our surveillance system also complies with the U.S. Tobacco Control Act and the FDA's draft guidance for modified risk tobacco product (MRTP) applications.
Find out more about PMI's safety surveillance system here.